Fenylketonúria - klasické príznaky, ako dedičná a diétna terapia
Obsah
- 1Ako sa manifestuje fenylketonúria
- 2Mechanizmus vývoja ochorenia
- 3Fenylketonúria u detí
- 4Symptómy choroby
- 5Príčiny a provokatívne faktory
- 6Diagnostika
- 7Liečba klasickej fenylketonúrie
- 8výživa novorodencov a diétna terapia
- 9Diéta pre deti predškolského veku a školákov
- 10Skupiny výrobkov pri PKU
- 11 , ako kontrolovať hladinu fenylalanínu v krvi
- 12Videá
, výskyt ochorení je spojené s genetickými defektmi v bunkovej stroje - fenylketonúriu - je zoznam niekoľkých dedičných chorôb na liečbu. Pioneer tohto ochorenia bol lekárom z Nórska, IA Fellynh neskôr zistilo, že rozvoj ochorenia a iba zodpovedné génu, tzv fenilalaninhidroksylazy genóm (chromozóm dlhé rameno 12oy obsahujúce až 4,5% z celkového materiálu bunkovej DNA). Dedičná defekt vedie k čiastočnej alebo úplnej inaktiváciu enzýmu fenylalanín pečene-4-hydroxylázy.
Ako sa manifestuje fenylketonúria
fenylketonúria dedičné ochorenie (PKU), vedie k chronickej otrave organizmus toxické látky, ktoré vznikajú v dôsledku poruchou metabolizmu aminokyselín a proceshydroxyláciou fenylalanínu. Konštantná intoxikácie spôsobuje poškodenie centrálneho nervového systému (CNS), ktorá pôsobí ako prejav postupnému poklesu inteligencie (fenylpyrovynohradnaya mentálna retardácia).
Fellingova choroba sa prejavuje nadmernou akumuláciou fenylalanínu v tele a produktmi jeho nevhodného metabolizmu. Medzi ďalšie faktory fenylketonúriu obavy transport aminokyselín cez BBB, nízkym počtom neurotransmiterov (serotonín, histamín, dopamín). Pri absencii včasnej liečby choroby vedie k mentálnej retardácii a môže spôsobiť smrť dieťaťa.

Mechanizmus vývoja ochorenia
Prychynoobrazuyuschym faktory genetických porúch metabolizmu je blok, ktorý zabraňuje tvorbe fenylalanín-4-hydroxylázy (enzým zodpovedný za premenu aminokyseliny tyrozínu v fenilaninu). Proteynohennaya aminokyselina tyrozín je zložkou proteínu a pigment melanín, ktoré sú dôležité pre fungovanie všetkých telesných systémov, a jeho nedostatok vedie k fermentopathy.
Inhibícia metabolitu spôsobená mutatívnou inaktiváciou enzýmu je aktivácia pomocných dráh na výmenu fenylalanínu. Aromatická alfa-aminokyselina v dôsledku chybných procesov výmeny sa rozkladá na toxické deriváty, ktoré sa nevytvárajú za normálnych podmienok:
- kyselina fenylpyruvátová (fenylpyruvát) - mastná aromatická alfa-ketoacid, jej vzdelanievedie k myelinizácii procesov neurónov a demencie;
- kyselina fenylmastná - produkt vytvorený počas regenerácie kyseliny fenyl-viriónovej;
- Fenyletylamín - počiatočná zlúčenina pre biologicky aktívne vysielače elektrochemických impulzov, zvyšuje koncentráciu dopamínu, adrenalínu a norepinefrínu;
- Ortofenylacetát - toxická látka, ktorá spôsobuje porušenie metabolických procesov fekálnych zlúčenín v mozgu.
Zo zdravotníckej štatistiky vyplýva, že patologicky pozmenený gén je prítomný v 2% populácie, ale neprejavuje sa. Genetická porucha sa prenáša na dieťa od rodičov iba v prítomnosti ochorenia u oboch partnerov, pričom dieťa sa v 50% prípadov stane nosičom mutovaného génu, zostáva zdravým. Pravdepodobnosť, že fenylketonúria u novorodencov spôsobí ochorenie, je 25%.
Akým typom sa zdedil
Škvrnková choroba je genetická odchýlka, na potlačenom autozomálnom recesívnom type. Tento typ dedičnosti znamená, že vývoj príznakov vrodených ochorení nastane, iba ak je jedno dieťa zdedené z chybnej genetickej kópie oboch rodičov, ktorí sú heterozygotnými nositeľmi zmeneného génu.
Vývin vrodených ochorení v 99% prípadov spôsobuje mutáciu génu zodpovedného za kódovanie enzýmov, ktorý poskytuje syntézu fenylalanínu-4-hydroxylázy (klasická fenylketonúria). Až 1% genetických ochorení súvisí s mutáciami, ktoré sa vyskytujú v iných génochnedostatočnosť dihydropteridínreduktázy (typ II PKU) alebo tetrahydrobiopterínu (PKU typu III).
Fenylketonúria u detí
Klasická forma genitálnej choroby u detí sa vo väčšine prípadov prejavuje zvonka viditeľnými príznakmi od 3-9 mesiacov života. Novorodenci s defektným génom vyzerajú zdravo, charakteristickým znakom je špecifický zvyk (vzhľad) dieťaťa. Symptóm sa zobrazí 6-12 mesiacov po narodení.
PKU typu II sa vyznačuje skutočnosťou, že prvé klinické príznaky sa objavujú po 1,5 rokoch od okamihu nástupu svetla. Príznaky ochorenia nezmiznú po diagnostikovaní genetických abnormalít a začiatku diétnej liečby. Tento typ vrodenej choroby často vedie k smrteľnému výsledku 2-3 rokov života dieťaťa. Najbežnejšie príznaky PKU typu II sú:
- vyjadrili odchýlky v duševnom vývoji;
- hyperreflexia;
- porušenie motorických funkcií všetkých končatín;
- syndróm nekontrolovaných svalových kontrakcií.
Klinické príznaky mutačných zmien v génoch typu III sú podobné ochoreniam vyskytujúcim sa u typu II. Nedostatok tetrahydrobiopterínu je charakterizovaný triadou špecifických príznakov:
- vysoký stupeň mentálnej retardácie;
- zdanlivo znížená veľkosť lebky vo vzťahu k iným častiam tela;
- spasticita svalov (s možnosťou úplnej straty pohybu končatín).

Vykazovanie Fellingovej choroby
V priebehu klinických štúdií a pozorovaní sa predpokladalo, že účinok boltoxický výmena deriváty fenylalanynovoho spôsobuje pokles mentálnych schopností, ktorý je progresívna a môže viesť k demencii (mentálna retardácia, hlúposť). Medzi možné príčiny ireverzibilné poruchy mozgovej činnosti považuje za primerané vzhľadom k nižším úrovniam tyrozín nedostatku neurotransmiterov, ktoré prenášajú impulzy medzi neurónmi.
Presná príčinná súvislosť medzi dedičné ochorenia a mozgové poruchy doteraz boli identifikované ako mechanizmu fenylketonúriou kvôli psychických stavov, ako je эhopraksyya, echolalia, záchvaty hnevu a podráždenosť. Výsledky analýz naznačujú, že fenylalanín má priamy toxický účinok na mozog, čo môže tiež spôsobiť zníženie inteligencie.
Štruktúra a fenotypové zvláštnosti
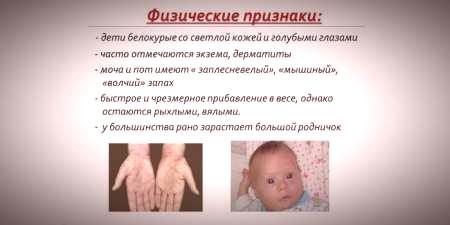
Vzhľadom na to, že sa nasýtenia pokožky a vlasov pigmentu závisí na úrovni tyrozínu v mitochondriách hepatocytov a spôsobuje fenylketonúriou zastavenie premenu fenylalanínu, u pacientov s týmto ochorením majú fenotypové vlastnosti (recesívne). Zvýšený svalový tonus spôsobuje výskyt odchýlok v štruktúre tela - stáva sa dysplastickou. Vynikajúce vonkajšie vlastnosti fenylketonúrie zahŕňajú:
- hypopigmentácia - ľahká pokožka, svetlé modré oči, odfarbené vlasy;
- cynizmus končatín;
- znížená veľkosť hlavy;
- zvláštnu pozíciu - keď sa snažia stať alebo sedieť dieťa vtelí "na mieru" (ruky a nohy ohnuté v kĺboch).
Symptómychoroba
Pri včasné odhalenie choroby Fellinha podstúpi úspešnú liečbu úpravou výživu a vývoj dieťaťa je v súlade s jeho vekovej skupine. Obtiažnosť pri zisťovaní génovej mutácie spočíva v tom, že skoré príznaky je ťažké odhaliť aj skúsený pediatr. Závažnosť symptómov vrodených chorôb sa zvyšuje s ich vekom dieťaťa, takže bielkovín v strave podporuje porúch centrálneho nervového systému.
Známky novorodencov
Počas prvých dní života známky patologických abnormalít ťažké odhaliť - dieťa vyzeralo prirodzené, vývojové oneskorenie pozorované. Príznaky ochorenia sa najprv objavia v 2 až 6 mesiacoch po narodení. Rodičia by mali byť upozornení správanie batoľa, ktorý sa vyznačuje nízkou aktivitu, letargia, alebo, sa týkajú hypervozbudymostyu.
Od začiatku dojčenia v tele novorodenca začína prúdiť s mliečnymi proteínmi, ktoré slúžia ako katalyzátor prvé príznaky, ktoré jasne ukazujú, že táto choroba začína pokrok. Špecifické klinické prejavy ochorenia zahŕňajú:
- trvalé vracanie (často sa používa na vrodené zúženie brankára);
- častá dislokácia;
- žiadna reakcia na vonkajšie podnety;
- svalová dystónia (znížené svalové napätie);
- konvulzívny syndróm (záchvaty epileptického alebo neepileptického typu).
Symptómy u detí po 6 mesiacoch
Ak prejav genetického ochorenia nie je(alebo nebola videná) počas prvých 6 mesiacov od narodenia dieťaťa, potom po tomto období môžete presne určiť oneskorenie v psychomotorickom vývoji. Príznaky genetických porúch spôsobených nedostatkom enzýmu u detí starších ako šesť mesiacov sú:
- zníženie aktivity (až do úplnej ľahostajnosti);
- nedostatok pokusov vstať, sedieť;
- špeciálna "myšia" vôňa kože (zápach plesní vzniká v dôsledku stiahnutia toxických derivátov fenylalanínu prostredníctvom potných žliaz a moču);
- strata schopnosti vizualizovať tváre rodičov;
- olupovanie kože;
- výskyt dermatitídy, ekzému, sklerodermy.
Progresia ochorenia v neprítomnosti liečby v detstve
Ak vývojové odchýlky neboli zistené v detstve a nebola vykonaná žiadna vhodná liečba, choroba sa začala aktívne rozvíjať a často vedie k postihnutiu. Nedostatok terapie v počiatočnom štádiu ochorenia spôsobuje vznik takých symptómov ochorenia vo veku 1,5 roka:
- mikrocefalie (znížená veľkosť mozgu);
- odtok (posun horného radu zubov dopredu);
- neskoršie vyrážanie zubov;
- hypoplázia skloviny (tenkosť alebo úplná absencia zubnej skloviny);
- oneskorenie jazykového vývoja až do úplného nedostatku prejavu;
- 3, 4 stupeň oligofrénie (mentálna retardácia, mentálna retardácia);
- vrodené srdcové ochorenie (poruchy štruktúry srdcového svalu, srdca,veľké plavidlá);
- poruchy vegetatívneho systému (akrocyanóza, zvýšené potenie, arteriálna hypotenzia);
- zápcha.
Príčiny a provokatívne faktory
Aby sa prejavila mutácia s autozomálnym recesívnym dedičstvom, musí byť dedičný gén zdedený oboma rodičmi. Genetické ochorenia tohto typu sa vyskytujú na rovnakej frekvencii u novorodencov a dievčat. Patogenéza PKU je spôsobená porušením výmeny fenylalanínu, ku ktorému dochádza v troch formách. Liečba diétnou terapiou podlieha klasickej fenylketonúrii typu I.
Atypické formy ochorenia možno liečiť úpravou výživy. Tieto odchýlky sú spôsobené nedostatkom tetrahydropteryna, dehydropterynreduktazы (zriedka - pyruvoyltetrahydropterynsyntazы, guanozín-5-tryfosfattsyklohydrolazы atď.). Väčšina prípadov smrteľných prípadov bola zaznamenaná u pacientov s zriedkavými zmenami v PKU, s klinickými prejavmi všetkých foriem ochorenia podobnými. Riziko pôrodu s mutovaným génom fenylalanínhydroxylázy sa zvyšuje, ak rodičia sú blízkymi príbuznými (s úzko súvisiacimi manželstvami).
Diagnostika
Podozrenie na genetické poruchy diagnostikované na základe súhrnných údajov získaných zo štúdia histórie ochorenia - genetických údajov, výsledkov klinického, lekárskeho a genetického výskumu. Na včasné zistenie vrodených ochorení (PKU, cystická fibróza, galaktozémia atď.) Program povinnej hmotnostiLaboratórne vyšetrenie všetkých novorodencov (neonatálny screening).
Ak si budúci rodičia uvedomujú prítomnosť mutovaného génu, moderná medicína ponúka spôsoby na zistenie poruchy počas tehotenstva (prenatálna diagnostika plodu invazívnou metódou). Na rozdelenie fenylketonúrie podľa druhu závažnosti sa používa podmienená klasifikácia založená na hladine fenylalanínu vo fibrinogénnej tekutine odvodenej z plazmatickej plazmy:
- Ťažká fenylketonúria - 1200 mkmol /l.
- Priemer - 60-1200 μmol /l.
- Jednoduché (bez liečby) - 480 μmol /l.
Skríningový test
Detekcia genetických abnormalít sa vyskytuje v niekoľkých štádiách. V prvej fáze v nemocnici u všetkých detí 3-5 dní života sa uskutočňuje odber periférnej krvi (z piatich) na výskum. Materiál sa aplikuje na papierovú formu a posiela sa do biochemickej laboratória, kde prebieha jej biochemická analýza. V druhom stupni skríningového testu sa stanoví koncentrácia fenylalanínu normálnej hodnoty.

Ak nie sú zistené žiadne patologické zmeny, diagnostika sa skončí, čo sa zaznamenáva na karte detí. Za prítomnosti odchýlok od normy sa výsledky diagnózy posielajú lekárovi-pediatrovi, aby poskytli podrobnú štúdiu krvnej vzorky novorodenca. Zdravie dieťaťa závisí od včasného a presného vykonania všetkých opatrení na zistenie abnormalít. Ak sa diagnóza potvrdí po opakovanom skríningovom vyšetrení, budú rodičia dieťaťaposlaný na kliniku pre detskú genetiku za účelom liečby.
Analýzy a štúdie na potvrdenie diagnózy
Opakovaná diagnostika v prípade zistenia odchýlky od normy počas počiatočnej skríningovej skúšky sa vykoná opätovným posúdením analýz. Okrem stanovenia obsahu fenylalanínu v krvi na metódy diagnostiky PKU u detí a dospelých patria:
- Test na ťažbu - stanovenie kyseliny fenylpyruvátovej v moči pridaním chloridu železa k biomateriálu (dochádza k modrozelenému zafarbeniu);
- Test Gatree - hodnotenie stupňa reakcie mikroorganizmov na metabolické produkty alebo enzýmy obsiahnuté v krvi pacienta;
- chromatografia - štúdium chemických vlastností látok rozdelených medzi dve fázy;
- fluorimetria - ožarovanie biomateriálu monochromatickým žiarením s cieľom určiť koncentráciu látok, ktoré sú v ňom obsiahnuté;
- elektroencefalografia - diagnostika elektrickej aktivity mozgu;
- zobrazovanie magnetickou rezonanciou - porušenie atómových jadier buniek elektromagnetickými vlnami a meranie ich odozvy.
Liečba klasickej fenylketonúrie
Základom terapie fenylketonúrie je obmedzenie spotreby produktov, ktoré sú zdrojom bielkovín živočíšneho a rastlinného pôvodu. Jediným spôsobom úspešnej liečby je diétna terapia, ktorej primeranosť sa odhaduje podľa obsahu fenylalanínu v sére. Maximálna prípustná hladina aminokyselín u pacientov rôznych vekových skupínje:
- u novorodencov a detí do 3 rokov - do 242 μmol /l;
- pre predškolákov - do 360 mkmol /l;
- u pacientov vo veku od 7 do 14 rokov - až do 480 μmol /l;
- u dospievajúcich - do 600 μmol /l.
Účinok stravy závisí od toho, v ktorom štádiu ochorenia ide o úpravu stravy. S včasnú diagnostiku vrodených vád stravy menovaný od 8 týždňov života (po uplynutí tejto doby sú nezvratné zmeny začínajú). Nedostatok včasné opatrenia vedie ku komplikáciám a nižšie IQ pri 4 ° C po dobu 1 mesiaca od narodenia do začiatku liečby.

Vzhľadom na to, že terapeutická strava pre fenylketonúriou poskytuje úplné vylúčenie zo stravy živočíšnych bielkovín, je potreba ďalších zdrojov esenciálnych aminokyselín a vitamínov B, vápnika - a fosfor obsahujúce minerálne zlúčeniny. Produkty, ktoré sa majú pridávať k nebielkovinovej diéte, zahŕňajú:
- proteínové hydrolyzáty (Amygens, Aminazol, Fibrinoses);
- neobsahujú fenylalanínové zmesi nasýtené esenciálnymi aminokyselinami - Tetrafen, bez fenylov.
Okrem terapeutických opatrení na riešenie príčin porušeniu fungovanie tela by mala byť symptomatická liečba zameraná na odstránenie vád a normalizáciu koordinácie reč motora. Kombinovaná terapia, vrátane fyzioterapie, masáže, logopedické služby, psychológov, vykonávať cviky. V niektorých prípadoch s dietnou terapiouukazuje použitie antikonvulzíva, nootropných a vaskulárnych liekov.
Vlastnosti liečby atypických foriem
fenylketonúria II a typ III neliečiteľné nyzkobelkovoy diéta - hladina fenylalanínu v krvi zostáva konštantný, zatiaľ čo pre obmedzenie prenosu proteínu v tele alebo klinických symptómov postupuje aj pri nižších úrovniach aminokyselín. Účinná liečba týchto foriem ochorenia sa uskutočňuje s použitím:
- tetrahydrobiopterín - faktor postihnutého enzýmu;
- syntetické analógy tetrahydrobiopterínu - tieto látky sa lepšie prenikajú cez hematoencefalickú bariéru;
- náhradná terapia lieky - neodstraňujú príčinu fenylketonúria, ale zachovať normálne fungovanie tela (levodopa spolu s Karbydofoy, oksytryptofan 5, 5 formyltetrahydrofolat);
- hepatoprotektory - podporujú fungovanie pečene;
- antikonvulzíva;
- zavedenie génu fenylalanínhydroxylázy do pečene - experimentálna metóda.
Funkcie výživy novorodencov a diétna terapia
V prvom roku života dieťaťa je povolené dojčiť, ale jeho množstvo by malo byť obmedzené. Od 6 mesiacov prijateľnú úroveň príjmu fenylalanínu je 60 až 90 mg na 1 kg telesnej hmotnosti dieťaťa (100 g mlieka obsahuje 5,6 mg fenylalanínu). Počnúc 3 mesiacmi by sa dieťa mala postupne rozširovať zavedením ovocných štiav a zemiakov.
Deti od 6 mesiacov umožnený vstup do stravy kaší zeleninu, obilniny (s sága) bezbilkovyhKiselev. Po 7 mesiacoch dieťa môže byť podávaný nyzkobilkovi cestoviny, 8 mesiacov - chlieb, ktorý obsahuje proteín. Age, ktorý by mal obmedziť tok bielkovín v tele chorého dieťaťa, nie je nainštalovaný. Lekári doteraz vykonaná diskusie o vhodnosti celoživotné diétu, ale súhlasí s tým, že minimálne 18 rokov, aby sa držať diétu.
fenylketonúria diagnostikované u žien nie je dôvod vzdať sa dieťa narodí. Tehotné ženy s PKU, aby nedošlo k poškodeniu plodu počas tehotenstva a je nutné pred plánovaným počatím a počas tehotenstva detskej stravy s obmedzením fenylalanínu prevencia možných komplikácií (jeho hladina v krvi by mala byť až 242 mmol /l).
Zmes laktózy pre deti
Diéta pre fenylketonúriou na základe zníženia podstatné dávky prirodzenej vlákniny v každodennej strave, ale telo novorodenca môže vyvíjať normálne pri absencii potrebných mikronutrientov. Pre naplnenie potrieb dieťaťa laktózy použitej v zmesi kyseliny proteín amino, ktorý, podľa ruského práva, musí byť k dispozícii u pacientov zdarma.
fenylalanín Tolerancia pre dieťa počas prvého roku života sa rýchlo mení a je potrebné kontrolovať jeho koncentrácia v krvi dieťaťa a vykonať úpravy v potrave. Zmesi určené pre vek:
- bol menovaný na deti Afenylak 15. Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- pre deti staršie ako 1rok predpísať obohatené vitamínmi a minerálmi zmes vysokého obsahu proteínov - PKU Prima, P-AM Univerzálna, PKU-1, PKU-2, HRM Maxamed, HR Maxumum.

Dietetické výrobky na doplnenie zásob bielkovín
Jednou z hlavných zložiek diétnej diéty pre fenylketonúriu sú nízkoproteínové potraviny na báze škrobu. Tieto doplnky obsahujú hydrolyzát kazeínu, tryptofánu, tyrozínu, metionínu, dusíka a zabezpečujú každodennú potrebu dieťaťa pre detský proteín, čo je nevyhnutné pre normálny vývoj a rast. Špecializované produkty, ktoré naplnia nedostatok potrebných minerálov a aminokyselín v nedostatku stravy, sú:
- Berlofen;
- Tsimorgan;
- Minafen;
- Apotoni.
Strava pre deti predškolského veku a školákov
Keď sa telo prispôsobuje fenylalanínu deťom od veku 5 rokov, je možné postupne znižovať obmedzenia týkajúce sa stravovania. Rozšírenie stravy je zavedením obilnín, mliečnych výrobkov, mäsových výrobkov. Študenti stredných škôl už majú vysokú toleranciu na fenylalanín, takže v tomto veku môžete pokračovať v rozširovaní stravy, zatiaľ čo je potrebné sledovať reakciu na všetky zmeny v strave. Nasledujúce metódy sa používajú na monitorovanie stavu dieťaťa:
- hodnotenie neurologických parametrov, psychologický stav;
- kontrola parametrov elektroencefalogramu;
- stanovenie hladiny fenylalanínu.
Skupiny výrobkov v PKU
V strave pacientov s PKU spolu s neproteínomŠkrobové výrobky a terapeutické zmesi zahŕňajú aj produkty prírodného pôvodu. Pri zostavovaní menu by ste mali jasne vypočítať množstvo konzumovaných bielkovín a neprekračovať odporúčaný lekár. Na vylúčenie toxických účinkov na telo 3 sa vytvorili zoznamy výrobkov, ktoré obsahujú zakázané (červené), nezastúpené (oranžové) a povolené (zelené) polohy.
Červený zoznam
Fenylketonúria sa vyvíja na pozadí neprítomnosti enzýmu, ktorý sa premieňa na tyrozín fenylalanín, takže vysoký obsah bielkovín je základom pre zaradenie produktov do zakázaného (červeného) zoznamu. Pozície z tohto zoznamu by mali úplne vylúčiť stravu pacienta na PKU:
- mäso;
- vnútorné orgány zvierat, vedľajšie produkty;
- klobásy, salámy;
- morské plody (vrátane rýb);
- vajcia všetkých vtákov;
- výrobky z kyslých mliečnych výrobkov;
- matice;
- zeleninové a obilné plodiny;
- sójové výrobky;
- želatinové jedlá;
- cukrovinky;
- aspartam.
Zoznam oranžových
Výrobky, ktoré sa majú podať do tela dieťaťa s diagnózou PKU, sú zaradené do oranžového zoznamu. Zahrnutie do zoznamu položiek z tohto zoznamu je prípustné, ale v prísne obmedzených množstvách. Hoci tieto produkty neobsahujú veľa bielkovín, ale môžem tiež zvýšiť hladinu fenylalanínu, preto ich použitie sa neodporúča:
- konzervovaná zelenina;
- zemiakové a ryžové jedlá;
- kapusta;
- mlieko;
- sherbet.
Zelený zoznam
Nebielkovinové produkty sú povolené na použitie u pacientov s fenylketonúriou bez akýchkoľvek obmedzení. Pred zakúpením tovaru zo zeleného zoznamu je potrebné preskúmať zloženie uvedené na obale a uistiť sa, že nie je prítomný fenylalanín obsahujúci aspartámové farbivo:
- ovocie;
- zelenina (okrem zemiakov a kapusty);
- bobule;
- zelené;
- škrobové obilniny (ságo);
- med, cukor, džem;
- výrobky z múky z kukurice alebo ryžovej múky;
- maslo, tuky (maslo, slnečnica, oliva).
Ako kontrolovať hladinu fenylalanínu v krvi
Fenylketonúria je nevyliečiteľná choroba, ktorá sa môže transformovať do stagnácie pomocou diétnej terapie a terapeutických a profylaktických opatrení. Ak dôjde k zmene životných podmienok, k porušeniu stravy, ochorenie sa môže znovu zhoršiť, takže pacienti potrebujú celoživotné pozorovanie. Kontrolným procesom je pravidelne stanovovať hladinu fenylalanínu v krvi. Frekvencia podávania závisí od veku pacienta:
- do 3 mesiacov - krvný skríning sa má vykonať týždenne, kým sa nedosiahnu stabilné výsledky;
- od 3 mesiacov do 1 roka - 1-2 krát za mesiac;
- od 1 do 3 rokov - 1 krát za 2 mesiace;
- za 3 roky - štvrťročne.

Krv na analýzu sa zdá byť 3-4 hodiny po jedle. Okrem skríningu sa vývoj PKU riadi stanovením nutričného stavu, fyzického, emočného vývoja pacienta, úrovne intelektuálnych schopnostía rozvoj jazyka. Podľa výsledkov pozorovaní môže byť potrebná ďalšia diagnostika za účasti príslušných odborníkov.
Videá
Informácie uvedené v článku sú informatívne. Materiály tohto článku nevyžadujú nezávislé zaobchádzanie. Len kvalifikovaný lekár môže diagnostikovať a poskytnúť poradenstvo o liečbe na základe individuálnych charakteristík konkrétneho pacienta.















